-
Featured News
Mayo Clinic Q and A: Diagnosing cystic fibrosis
Share this:




 DEAR MAYO CLINIC: Recently, my niece was diagnosed with cystic fibrosis at 16. No one else in our family has been diagnosed with it, and, until recently, she didn’t have any symptoms. Now I’m worried about my kids, ages 4 and 6. Their newborn screenings for cystic fibrosis were negative, but should I have them tested anyway?
DEAR MAYO CLINIC: Recently, my niece was diagnosed with cystic fibrosis at 16. No one else in our family has been diagnosed with it, and, until recently, she didn’t have any symptoms. Now I’m worried about my kids, ages 4 and 6. Their newborn screenings for cystic fibrosis were negative, but should I have them tested anyway?
ANSWER: Because of the way cystic fibrosis is passed from parents to children, one member of an extended family may be affected, while many others are not. If your children are healthy, without any signs of the disease, it’s unlikely they will develop cystic fibrosis, especially because their newborn screenings were negative. However, the test for cystic fibrosis is a simple, noninvasive and inexpensive test. If it’s negative, that would rule out cystic fibrosis in your children. So it may be beneficial to have that test performed.
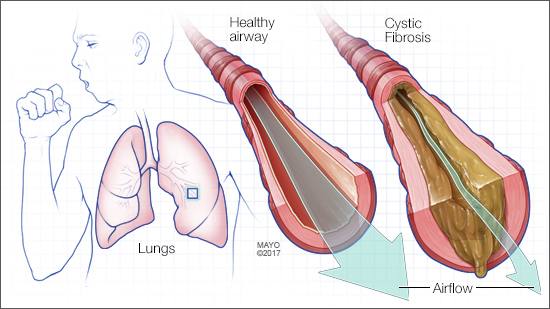
Cystic fibrosis is a disease affecting the cells that produce mucus, sweat and digestive juices. These fluids are normally thin and slippery. In people with cystic fibrosis, they become sticky and thick. Instead of acting as a lubricant, the secretions plug up tubes, ducts and passageways, especially in the lungs and pancreas.
Signs and symptoms of cystic fibrosis are different, depending on the severity of the disease. Because cystic fibrosis affects the lungs, common symptoms include a chronic cough and frequent pneumonia. The disease also can affect the gastrointestinal tract, causing bulky, greasy stools and poor growth. Although unusual, some people don’t notice symptoms until they are teenagers or adults, as in your niece’s case.
An inherited disease, cystic fibrosis happens due to a genetic problem. A mutation in a gene changes a protein that regulates the movement of salt in and out of cells. To develop cystic fibrosis, though, a child has to inherit two copies of the defective gene — one from each parent. If children inherit only one copy, they won’t develop cystic fibrosis. But with one defective gene, they are considered cystic fibrosis carriers and could pass the genetic defect to their own children.
Newborn screening for cystic fibrosis is now performed in every state in the U.S. As a result, the condition often can be identified within the first month of life — before symptoms start. That’s important because proper nutrition, including vitamins and enzymes to help digest food, may help lessen symptoms in children who are diagnosed early.
The screening involves checking a blood sample for elevated levels of a chemical called immunoreactive trypsinogen. Because cystic fibrosis is not the only reason the level of this chemical may be high, if the results of the screening are positive, more testing is needed to confirm a diagnosis. Health care providers also may recommend genetic tests to identify specific defects on the gene responsible for cystic fibrosis.
In your niece’s case, she may have been born before newborn screening for cystic fibrosis began, depending on where she lived. Or, because the newborn test for cystic fibrosis is a screening test, occasionally it doesn’t identify the disease in a child. It’s always recommended — even if a child has a negative newborn screen — to repeat testing for cystic fibrosis if the disease is suspected.
In many children with a positive newborn screening, further testing reveals they are carriers of a mutation for cystic fibrosis but not at risk to develop the disease. The test used to identify cystic fibrosis is a sweat test. For this test, a chemical that produces sweat is applied to a small area of skin on each arm. Then, the sweat is evaluated to see if it’s saltier than normal.
If you are interested in having your children tested for cystic fibrosis, talk with their health care provider. He or she can review your children’s health history and genetics to help you decide whether the testing may be appropriate. — Dr. Julie Baughn, Pediatric Pulmonology, Mayo Clinic, Rochester, Minnesota


