-
Cardiovascular
Mayo Clinic Q and A: Understanding and treating long QT syndrome
Share this:




 DEAR MAYO CLINIC: I recently read that long QT syndrome is quite common. What is it, and how is it diagnosed? I have read that fainting may be one sign of the disorder. Can long QT syndrome be treated?
DEAR MAYO CLINIC: I recently read that long QT syndrome is quite common. What is it, and how is it diagnosed? I have read that fainting may be one sign of the disorder. Can long QT syndrome be treated?
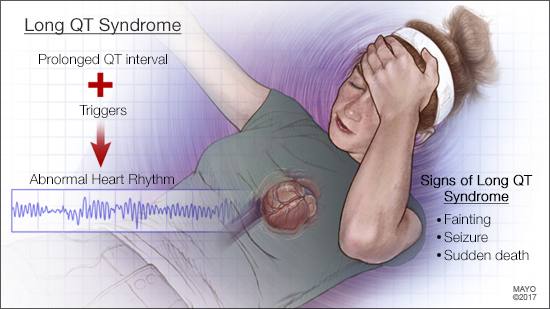
ANSWER: Long QT syndrome is a heart rhythm disorder that is diagnosed due to symptoms, an abnormal 12-lead electrocardiogram (ECG), or as a result of a family history of the disease. Once it’s diagnosed, long QT syndrome often can be treated effectively.
Your heart circulates blood throughout your body each time it beats. The chambers of your heart contract and relax to pump the blood. These actions are controlled by electrical impulses that travel through your heart. After each heartbeat, your heart’s electrical system recharges itself in preparation for the next heartbeat.
In long QT syndrome, the heart muscle takes longer than normal to electrically recharge after it contracts. A test called an electrocardiogram (also called an ECG or EKG) can detect this problem. Most people don’t have routine ECGs, though, so catching it with this test if you haven’t had any symptoms, or if you don’t have a family history of the disorder, is less common.
Congenital or inherited long QT syndrome affects about 1 in 2,000 people. That statistic, however, is likely an underestimate, because people can have the disorder for years and not know it. In addition, some unexplained deaths, such as those by drowning or unexplained vehicle accidents, may be the result of sudden cardiac arrest triggered by long QT syndrome.
As you note, one of the most frequent symptoms of long QT syndrome is fainting. The disorder triggers fainting when the heartbeat becomes irregular — usually during exercise or other physical exertion, or when you become scared, excited or angry. If the heartbeat continues to be dangerously fast and erratic for a prolonged period of time, long QT syndrome also can lead to seizures due to lack of oxygen to the brain. If the heartbeat doesn’t return to normal, sudden death can occur.
At this time, universal screening does not include long QT syndrome. In most cases, it’s diagnosed with an ECG after someone experiences a warning symptom — usually fainting without warning. Because long QT syndrome often has a genetic component, some people may be diagnosed if a family member has the disorder. Or, in some cases, first-degree family members (parents, siblings and children) may be tested for long QT syndrome when there is an unexplained, sudden death in a family. Genetic testing should be recommended as part of evaluation for long QT syndrome.
Once long QT syndrome is diagnosed, treatment usually starts with a medication called a beta-blocker. Other medications to counteract irregular heart rhythm may be used in conjunction with beta-blockers.
If medication isn’t enough to control long QT, a surgery called left cardiac sympathetic denervation may be recommended. During this minimally invasive procedure, nerves that release a chemical called noradrenalin onto the heart’s muscle are removed from along the left side of the spine. The surgery significantly reduces the risk of a long QT-triggered faint, seizure or sudden death.
Sometimes, despite medications and/or the denervation surgery, a person with long QT syndrome may be at unacceptably high risk for sudden death. In those situations, a defibrillator may be implanted under the skin of the chest. The device can stop a potentially fatal irregular heart rhythm by delivering an electric shock to reset the rhythm back to normal.
Treatment for long QT syndrome is tailored to the individual after a comprehensive risk assessment. In most cases, once it’s identified, long QT syndrome can be treated successfully so the patient should be able to live and thrive despite the diagnosis.
Given the complexity of this disease and the importance of getting the diagnosis and treatment plan right, people suspected of having long QT syndrome should be referred to a center of excellence dedicated to families with this potentially lethal, yet highly treatable, genetic heart disease. — Dr. Michael J. Ackerman, Cardiovascular Diseases, Mayo Clinic, Rochester, Minnesota


